Publications
2023
“The Role of the Transient Atropisomerism and Chirality of Flurbiprofen Unveiled by Laser-Ablation Rotational Spectroscopy”
Andrés Verde, Juan Carlos López, and Susana Blanco*
Chem. Eur. J. 2023, e202300064 (1 of 6). DOI: 10.1002/chem.202300064
The combination of atropisomerism and chirality in flurbiprofen is shown to be relevant concerning its pharmacological activity. The two most stable conformers of a total of eight theoretically predicted for each R- or S- flurbiprofen enantiomers have been isolated in the cooling conditions of a supersonic jet and structurally characterized by laser ablation Fourier transform microwave spectroscopy. The detected conformers, whose structure is mainly defined by three dihedral angles, only differ in the sign of the phenyl torsion angle giving rise to Sa and Ra atropisomers. A comparison with the structures available for the R- and S- enantiomers complexed to COX isoforms reveals that the enzymes select only the Sa atropisomers, resulting in a diastereoisomerspecific recognition. The most stable gas phase conformer is exclusively selected when using the S- enantiomer while the
second is recognized only for the R- enantiomer. These experimental results highlight the importance of atropisomerism in drug design.
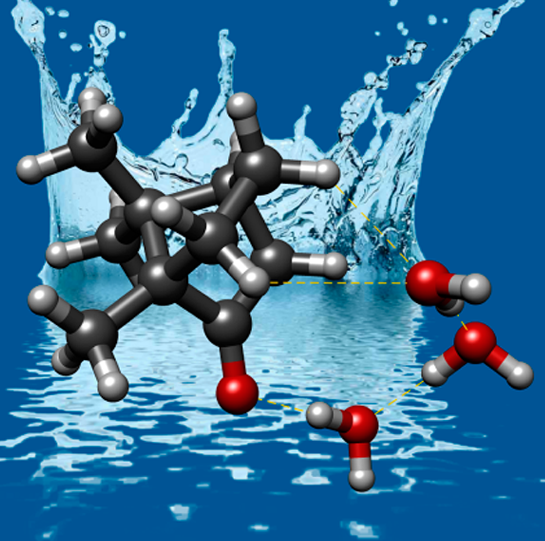
“Quantum Tunneling Facilitates Water Motion across the Surface of
Phenanthreneal”
Donatella Loru, Amanda L. Steber, Cristóbal Pérez, Daniel A. Obenchain, Berhane Temelso,
Juan C. López, and Melanie Schnell*
J. Am. Chem. Soc. 2023, 145, 17201−17210. DOI: 10.1021/jacs.3c04281

2022
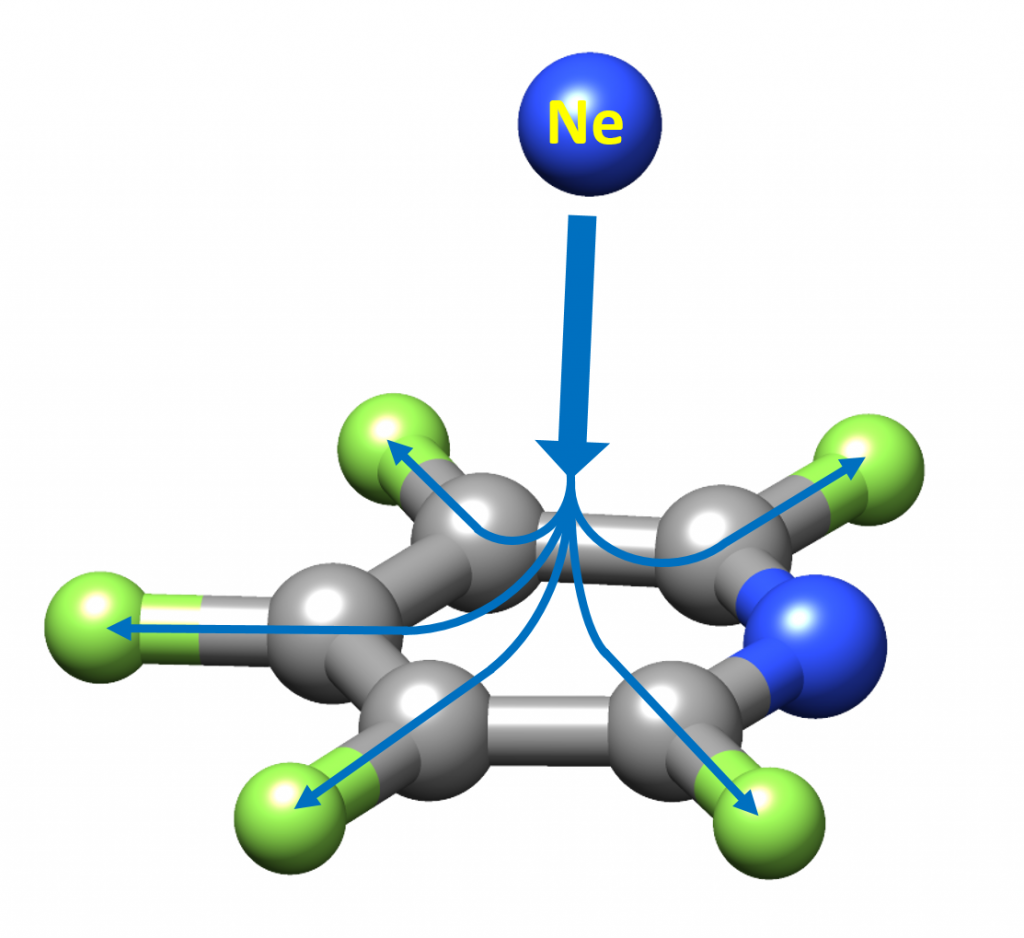
“Perfluorination of Aromatic Compounds Reinforce Their van der Waals Interactions with Rare Gases: The Rotational Spectrum of Pentafluoropyridine-Ne”
Alberto Macario, Susana Blanco, Ibon Alkorta and Juan Carlos López*
Molecules 2021, 26, 7585. DOI: 10.3390/molecules27010017
2021
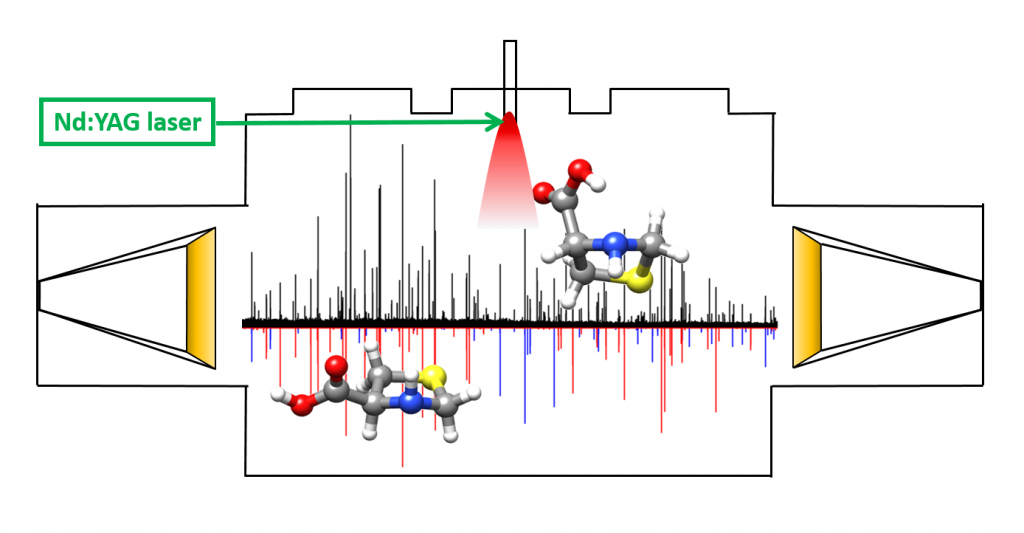
“Decoding the Structure of Non-Proteinogenic Amino Acids:
The Rotational Spectrum of Jet-Cooled Laser-Ablated Thioproline”
Juan Carlos López, Alberto Macario, Andrés Verde, Alfonso Pérez-Encabo and Susana Blanco*
Molecules 2021, 26, 7585. DOI: 10.3390/molecules26247585
“How Aromatic Fluorination Exchanges the Interaction Role of Pyridine with Carbonyl Compounds: The Formaldehyde Adduct”
Juan Carlos López,* Alberto Macario, Assimo Maris, Ibon Alkorta, and Susana Blanco*
Chem. Eur. J. 2021, 27, 13870–13878. DOI: 10.1002/chem.202102163
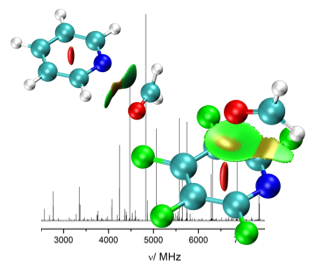
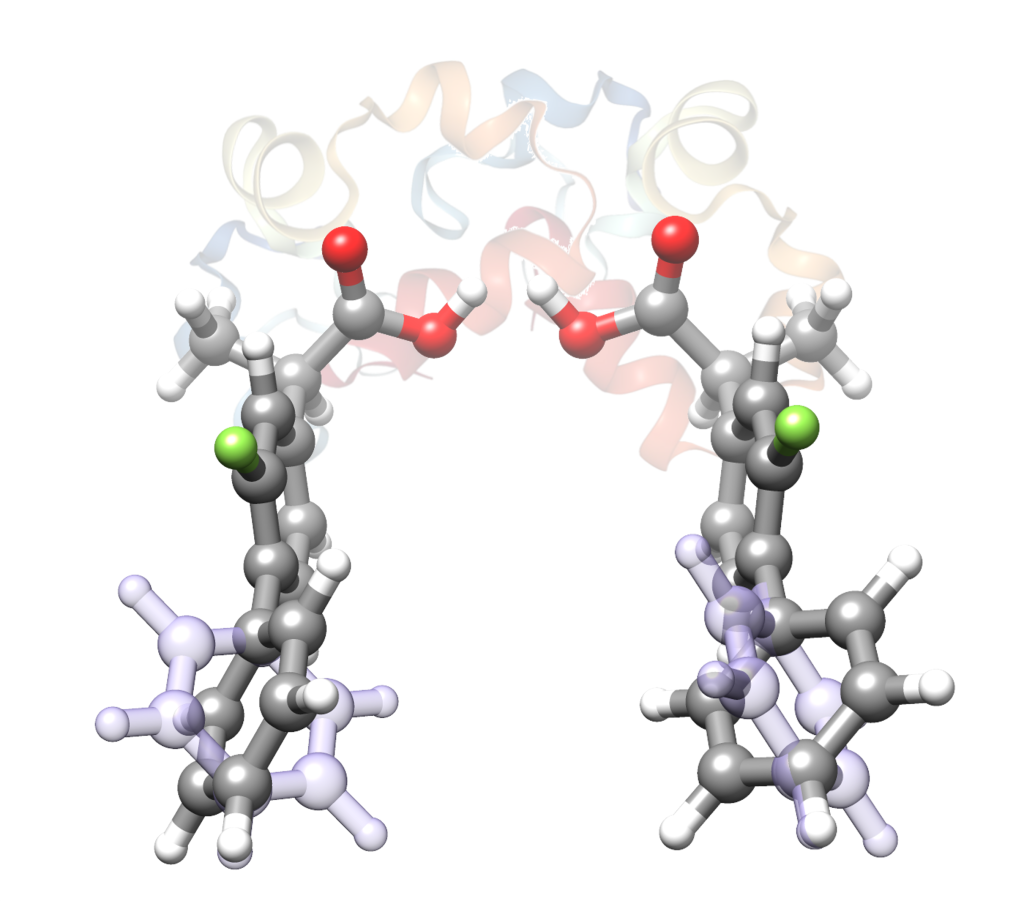
“The structure of isolated thalidomide as reference for its chirality-dependent biological activity: a laser-ablation rotational study“
S. Blanco,* A. Macario, J. C. López*
Phys. Chem. Chem. Phys., 23, (2021), 13705-13713. DOI: 10.1039/D1CP01691A
Thalidomide is a drug that presents two enantiomers with markedly different pharmacological and toxicological activities. It is sadly famous due to its teratogenic effects mostly caused by the preferential docking of the (S)-enantiomer to the target protein cereblon (CRBN). To compare the structure of the bound CRBN thalidomide enantiomers with that of the isolated molecule, the rotational spectrum of laser-ablated thalidomide has been studied by chirp-pulsed Fourier transform microwave spectroscopy in supersonic jets complemented by theoretical computations. A new setup of the laser ablation nozzle used is presented. Two stable equatorial and axial conformers of thalidomide have been predicted corresponding to the two possible bent conformations exhibited by the glutarimide moiety. Only the most stable equatorial conformer has been detected. The comparison of its structure with those of the (S)- and (R)-enantiomers bound to CBRN shows that the bound (S) species is only slightly distorted. On the contrary, the bound (R)-enantiomer exhibits a highly distorted structure which affects the degree of puckering of the glutarimide ring and especially to the orientation of the phtalimide and glutarimide subunits. This is consistent with a less stable (R)-enantiomer and the known preference of (S)-thalidomide to bind CRBN, which starts the process leading to teratogenic effects.
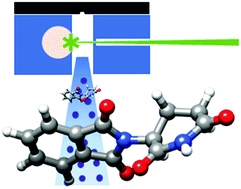
“Microwave detection of wet triacetone triperoxide (TATP): non-covalent forces in water dynamics“
S. Blanco, A. Macario, J. García-Calvo, A. Revilla-Cuesta, T. Torroba, J. C. López*
Chem. Eur. J., 27, (2021), 1680-1687. DOI: 10.1002/chem.202003499

The water adducts of triacetone triperoxide (TATP) have been observed by using broadband rotational spectroscopy. This work opens a new way for the gas‐phase detection of this improvised explosive. The observed clusters exhibit unusual water dynamics and rarely observed multicenter interactions. TATP‐H2O is formed from the D3 symmetry conformer of TATP with water lying close to the C3 axis. Water rotation around this axis with a very low barrier gives rise to the rotational spectrum of a symmetric top. The main interaction of the monohydrate is a four‐center trifurcated donor Ow‐H⋅⋅⋅O hydrogen bond, not observed previously in the gas phase, reinforced by a weak four‐center trifurcated acceptor C−H⋅⋅⋅Ow interaction. Surprisingly, all structural signatures show the weakness of these interactions. The complex TATP‐(H2O)2 is formed from the monohydrated TATP by the self‐association of water.
2020
“Microsolvation of ethyl carbamate conformers: effect of carrier fas on the formation of complexes“
P. Pinacho, J. C. López,* Z. Kisiel, S. Blanco*
Phys. Chem. Chem. Phys., 22, (2020), 18351-18360. DOI: 10.1039/d0cp03093g
Microsolvated complexes of ethyl carbamate (urethane) with up to three water molecules formed in a supersonic expansion have been characterized by high-resolution microwave spectroscopy. Both chirped-pulse and cavity Fourier transform microwave spectrometers covering the 2–13 GHz frequency range have been used. The structures of the complexes have been characterized and show water molecules closing sequential cycles through hydrogen bonding with the amide group. As is the case in the monomer, the ethyl carbamate–water complexes exhibit a conformational equilibrium between two conformers close in energy. The interconversion barrier between both forms has been studied by analyzing the spectra obtained using different carrier gas in the expansion. Complexation of ethyl carbamate with water molecules does not appear to significantly alter the potential energy function for the interconversion between the two conformations of ethyl carbamate.
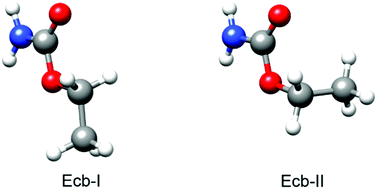
“Terpenoids: shape and non-covalent interactions. The rotational spectrum of cis-verbenol and its 1:1 water complex“
S. Blanco, J. C. López, A. Maris*
Phys. Chem. Chem. Phys., 22, (2020), 5729-5734. DOI: 10.1039/d0cp00086h
Verbenols are aromatic terpenoids whose bioactivity is attracting considerable experimental efforts. Exploiting the chirped-pulse Fourier transform technique, the rotational spectra of cis-verbenol, its hydroxyl deuterated form, and all 13C-monosubstituted isotopologues have been assigned, allowing for the structure determination, as the knowledge of its shape is crucial to understanding its molecular activity. Unlike in the solid state, in the gas phase, the most stable conformer exhibits an anti HO–CH arrangement, analogous to that of simpler allyl alcohol compounds. Observation of the 1 : 1 water complex showed that the conformation of cis-verbenol is still anti where water not only acts mainly as a proton donor to the hydroxyl group, but also as a proton acceptor, forming a secondary C–H⋯O interaction with the hydrogen atom of alkyl verbenol.
2019
“Pyridine-acetaldehyde, a molecular balance to explore the n→π* interaction“
S. Blanco,* A. Macario, J. C. López*
Phys. Chem. Chem. Phys., 21, (2019), 20566-20570. DOI: 10.1039/c9cp04088a
The complex pyridine–acetaldehyde is formed through an n-π* interaction and a C–H···O contact. The acetaldehyde methyl group internal rotation induces a phase-locked intermolecular oscillation along the Burgi–Dunitz coordinate. Surprisingly, this sort of molecular balance extracts energy through the n-π* interaction to reduce the size of the internal rotation barrier.
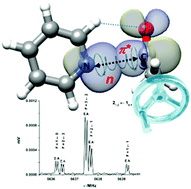
“Competition between intra- and intermolecular hydrogen bonding: o-anisic acid···formic acid heterodimer”
A. Macario, S. Blanco,* J.Thomas, Y. Xu, J. C. López*
Chem. Eur. J., 25, (2019), 12325-12331. DOI: 10.1002/chem.201902086

Four conformers of the heterodimer o-anisic acid–formic acid, formed in a supersonic expansion, have been probed by Fourier transform microwave spectroscopy. Two of these forms have the typical double intermolecular hydrogen-bond cyclic structure. The other two show the o-anisic acid moiety bearing a trans-COOH arrangement supported by an intramolecular O-H···O bond to the neighbor methoxy group. In these conformers, formic acid interacts with o-anisic acid mainly through an intermolecular O-H···O hydrogen bond either to the O-H or to the C=O moieties, reinforced by other weak interactions. Surprisingly, the most abundant conformer in the supersonic expansion is the complex in which the o-anisic acid is in trans arrangement with the formic acid interacting with the O-H group. Such a trans-COOH arrangement in which the intramolecular hydrogen bond dominates over the usually observed double intermolecular hydrogen bond interaction has never been observed previously in an acid–acid dimer.
“Conformational landscape of m-anisic acid and its complexes with formic acid”
A. Macario, S. Blanco,* J.Thomas, Y. Xu, J. C. López*
J. Phys. Chem. A, 123, (2019), 6772-6780. DOI: 10.1021/acs.jpca.9b05295
Four conformers of m-anisic acid were observed in a supersonic expansion using Fourier transform microwave spectroscopy. These conformers correspond to different relative orientations of the acid and methoxy groups and have their planar skeletons stabilized by resonance. When formic acid was present in the jet, the spectra of four m-anisic acid−formic acid heterodimer conformers were detected. The complexes are formed from the interaction of formic acid with each of the four observed conformers of m-anisic acid through the typical acid−acid sequential cycle with a double O−H···O−C hydrogen bond interaction in a pseudo eightmemberedring arrangement. The four heterodimer forms retain the same four m-anisic acid conformational geometries and the same relative abundances of the m-anisic acid monomeric forms in the supersonic expansion. This proves that a kinetic mechanism dominates the formation of complexes in the jet.
“Structure of butyl carbamate and of its water complex in the gas phase”
P. Pinacho, J. C. López,* Z. Kisiel, S. Blanco*
J. Phys. Chem. A, 123, (2019), 7983-7990. DOI: 10.1021/acs.jpca.9b06638
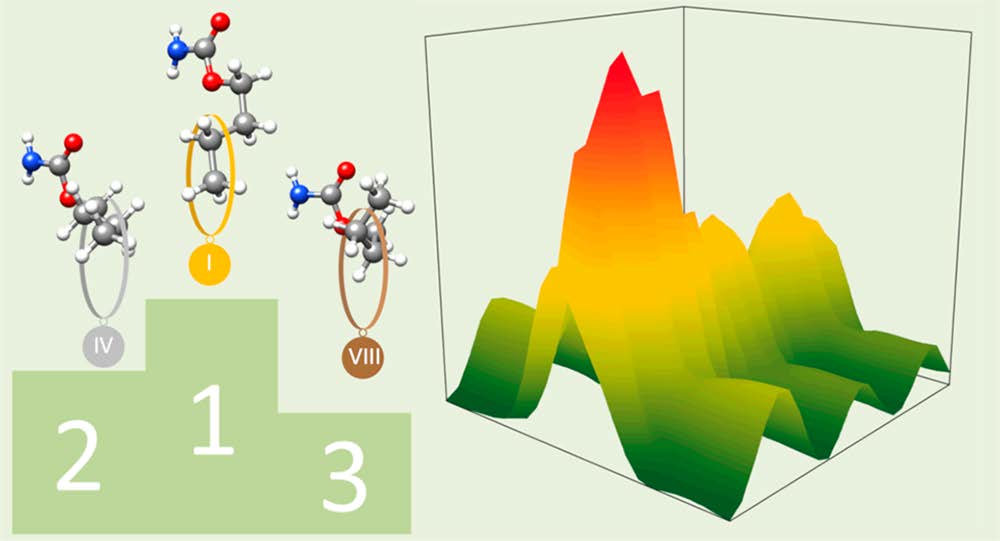
The structure of butyl carbamate and of its complex with water generated in a supersonic expansion has been characterized by Fourier transform microwave spectroscopy. Up to 13 low-energy conformations of the monomer have been predicted that differ in the relative orientation of the butyl chain and the amide group. However, only three
conformations have been observed experimentally. The remaining low-energy conformers are expected to interconvert into the observed rotamers through collisional relaxation processes in the supersonic jet. The values of the C-O-Cα-Cβ dihedral angle observed for the two most stable conformers of butyl carbamate, with extended configurations, can be directly correlated with the values of this angle in the two experimentally observed conformers of the shorter-chain molecule, ethyl carbamate. The less stable form shows a weak C-H···O-C intramolecular hydrogen bond from the terminal methyl group to the carbamate C-O group, stabilizing a folded configuration. For the most stable butyl carbamate monomer the complex with one molecule of water has been observed. In that complex the water molecule attaches to the amide group in a cyclic arrangement using two hydrogen bonds. The results indicate that water does not substantially alter the conformational behavior of butyl carbamate.
“Conformational equilibria in o-anisic acid and its monohydrated complex: the prevalence of the trans-COOH form”
J. C. López,* A. Macario, S. Blanco*
Phys. Chem. Chem. Phys., 21, (2019), 6844-6850. DOI: 10.1039/c9cp00552h
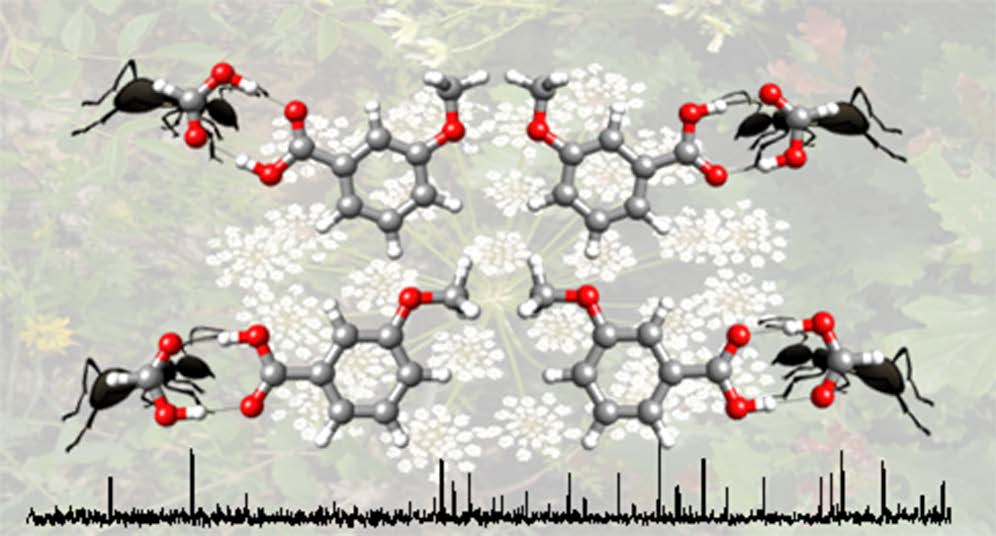
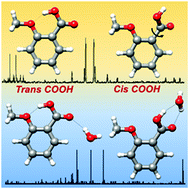
The conformation of ortho-anisic acid and its monohydrated clusters generated in a supersonic jet has been studied by rotational spectroscopy to analyze the structural implications of ortho substitution. The dominant monomer species shows a trans-COOH arrangement stabilized by an intramolecular O–H···O hydrogen bond from the acid to the methoxy group. The spectra of two non-planar skeleton monomer forms with a cis-COOH arrangement have also been detected. A tunneling doublet has been observed for the most stable cis-COOH conformer. The periodic potential energy function for the COOH internal rotation connecting these cis forms has been estimated from the experimental data. For the first time a water complex with an acid in a trans-COOH configuration has been observed. This is more abundant than the other weak form observed for the complex, characterized by a cis-COOH arrangement. The observation of the rotational spectra of salicylic acid, methyl salicylate and methyl 2-methoxybenzoate, formed upon heating of ortho-anisic acid, gives additional interest in the chemistry of ortho substituted benzoic acids.
“The complete conformational panorama of formanilide-water complexes: the role of water as a conformational switch”
P. Pinacho, S. Blanco,* J. C. López*
Phys. Chem. Chem. Phys., 21, (2019), 2177-2185. DOI: 10.1039/c8cp06959j
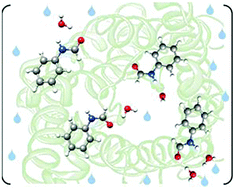
The microsolvated complexes of formanilide, generated in a supersonic expansion, have been observed by Fourier transform microwave spectroscopy. Three 1 : 1 and one 1 : 2 formanilide–water adducts have been observed and their structures characterized by the measurement of isotopologue rotational spectra. In one of the monohydrated complexes and in the dihydrated complex, formanilide adopts a cis-configuration. In these species water closes sequential cycles with the cis amino and carbonyl groups through a network of N–H···O and O–H···O hydrogen bonds. Furthermore, in these complexes cis-formanilide has almost the same non planar configuration observed in the monomer. In the two monohydrated complexes detected with trans-formanilide, a planar skeleton is detected with water interacting solely with either the amino (N–H··O bond) or the carbonyl group (O–H···O=C bond). The observed tunnelling splittings show a rich intermolecular dynamics in those complexes. The results seem to indicate that complexation with water switches the configuration of formanilide from trans, which is more stable for the bare monomer, to cis, which is more stable for the hydrated complexes.
“Water induces the same crown shapes as Li+ or Na+ in 15-crown-5 ether: a broadband rotational study”
J. C. López,* C. Pérez, S. Blanco, V. A. Shubert, B. Temelso, G. C. Shields, M. Schnell
Phys. Chem. Chem. Phys., 21, (2019), 2875-2881. DOI: 10.1039/C8CP05552A
15-Crown-5 ether (15C5) and its complexes with water have been studied using broadband Fourier transform microwave spectroscopy in a supersonic jet. A new conformer of 15C5 has been observed and established as the new global minimum out of a total of nine isolated structures. In addition, two 15C5–H2O and two 15C5–(H2O)2 clusters have been observed. The cluster structures have been unambiguously identified through the observation of water 18O isotopologue spectra. In all the clusters, at least one water molecule, located close to the axis of the 15C5 ring, interacts through two simultaneous hydrogen bonds to the endocyclic oxygen atoms. This interaction reshapes the 15C5 ring to reduce its rich conformational landscape to only two open structures, related to those found in complexes with Li+ or Na+ ions. In the most abundant 15C5–(H2O)2 form, the two water molecules repeat the same interaction scheme while binding to opposite sides of the ring. In the second most abundant dihydrated form the two water molecules lie on the same side of the ring. This finding is exceptionally rare because water–water interactions typically prevail over the formation of additional solute-water contacts, and it showcases the particular binding features of crown ethers.

2018
“Rotational Characterization of an n→π* Interaction in a Pyridine-Formaldehyde Adduct”
S. Blanco, J.C. López*
J. Phys. Chem. Letts., 9, (2018), 4632-4637. DOI: 10.1021/acs.jpclett.8b01719
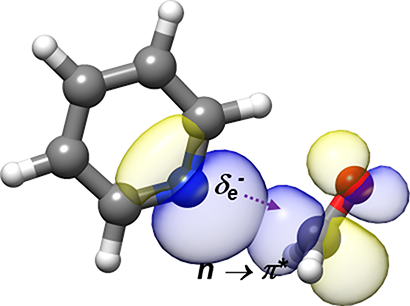
The rotational spectrum of the pyridine−formaldehyde adduct generated in a supersonic expansion has been analyzed using Fourier transform microwave spectroscopy. The spectrum shows a quadrupole coupling hyperfine structure due to the presence of 14N. The spectra of the parent species, 13C and 15N isotopologues, have been observed to investigate its structure. The complex shows Cs symmetry with the plane of pyridine bisecting the ∠HCH angle of formaldehyde and the N atom located along the Bürgi−Dunitz trajectory of nucleophile addition to a carbonyl group (r(N−C) = 2.855(4) Å, ∠NC=O = 102.8(6)°). From this structure and with the help of ab initio computations and natural bond orbital analysis, it is shown unambiguously that pyridine links to formaldehyde, the smallest molecule bearing a carbonyl group, through an n → π* interaction together with a weak C−H···O bond.
“Microsolvated complexes of ibuprofen as revealed by high-resolution rotational spectroscopy”
P. Pinacho, A. Krin, C. Pérez, S. Zinn, J.C. López, S. Blanco*, M. Schnell*
Phys. Chem. Chem. Phys., 20, (2018), 15635-15640. DOI: 10.1039/C8CP01088A
Hydrogen-bonded complexes between ibuprofen and water generated in a supersonic expansion were characterized using chirped-pulse Fourier transform microwave spectroscopy in the 2–8 GHz frequency range. Four spectra were observed allowing the determination of their rotational parameters. Comparison with quantum-chemical calculations led to their identification as the lowest energy 1 : 1 ibuprofen–water complexes. These correspond to the complexes between water and the four different conformers of ibuprofen previously detected in the gas phase, owing to their similar stabilization energies and abundances. Water seems to not change the conformational distribution of ibuprofen.

“Structure Determination, Conformational Flexibility, Internal
Dynamics, and Chiral Analysis of Pulegone and Its Complex with
Water”
A. Krin, C. Pérez*, P. Pinacho, M.M. Quesada-Moreno, J.J. López-González, J.R. Avilés-Moreno, S. Blanco, J.C. López, M. Schnell*
Chem. Eur. J., 24, (2018), 721-729. DOI:10.1002/chem.201704644
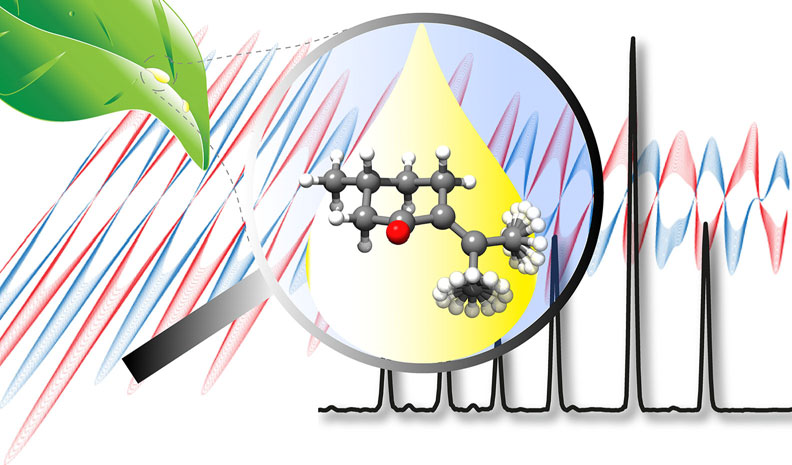
In the current work we present a detailed analysis of the chiral molecule pulegone, which is a constituent of essential oils, using broadband rotational spectroscopy. Two conformers are observed under the cold conditions of a molecular jet. We report an accurate experimentally determined structure for the lowest energy conformer. For both conformers, a characteristic splitting pattern is observed in the spectrum, resulting from the internal rotation of the two non-equivalent methyl groups situated in the isopropylidene side chain. The determined energy barriers are 1.961911(46) kJmol1 and 6.3617(12) kJmol1 for one conformer, and 1.96094(74) kJmol1 and 6.705(44) kJmol1 for the other one. Moreover, a cluster of the lowest energy conformer with one water molecule is reported. The water molecule locks one of the methyl groups by means of a hydrogen bond and some secondary interactions, so that we only observe internal rotation splittings from the other methyl group with an internal rotation barrier of 2.01013(38) kJmol1. Additionally, the chirality-sensitive microwave three-wave mixing technique is applied for the differentiation between the enantiomers, which can become of further use for the analysis of essential oils.
2017
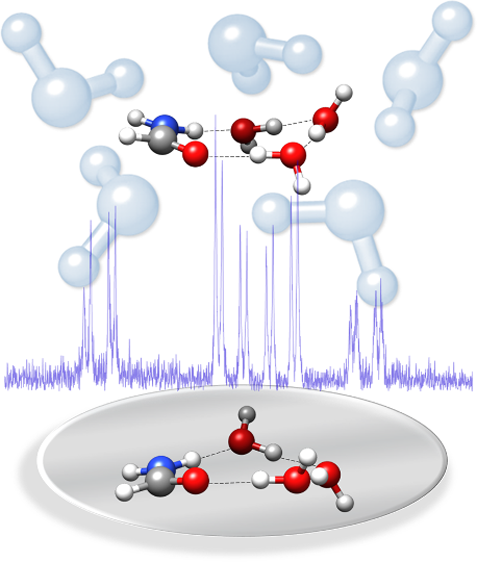
“Structure and Dynamics in Formamide−(H2O)3: A Water Pentamer Analogue”
S. Blanco*, P. Pinacho, J.C. López*
J. Phys. Chem. Letts., 8, (2017), 6060-6066. DOI: 10.1021/acs.jpclett.7b02948
Water self-association dominates the formation of microsolvated molecular clusters which may give rise to complex structures resembling those of pure water clusters. We present a rotational study of the complex formamide−(H2O)3 formed in a supersonic jet and several monosubstituted isotopologues. Formamide and water molecules form a four-body sequential cycle through N−H···O, O−H···O, and O−H···O=C hydrogen bonds, resulting in a chiral structure with a nonplanar skeleton that can be overlapped to that of water pentamer. The analysis of the 14N-nucleus quadrupole coupling effects shows the depletion of the electron density of the N atom lone pair with respect to the bare formamide that affects the amide group C−N and C=O distances. The study of the observed tunneling doublets shows that formamide−(H2O)3 follows a path to invert its structure driven by the flipping of water subunits and passing through successive nonplanar configurations, a motion reminiscent of the pseudorotation of water pentamer.
“Corannulene and its complex with water: a tiny
cup of water “
C. Pérez, A.L. Steber, A.M. Rijs, B. Temelso, G.C. Shields, J.C. López, Z. Kisiel*, M. Schnell*
Phys. Chem. Chem. Phys., 19, (2017), 14214-14223. DOI:10.1039/c7cp01506b

We report the results of a broadband rotational spectroscopic study of corannulene, C20H10, all of its singly substituted 13C isotopologues, and a complex of corannulene with one molecule of water. Corannulene is a polycyclic aromatic hydrocarbon (PAH) with a curved structure that results in a large dipole moment. Observation of 13C isotopic species in natural abundance allowed us to precisely determine the molecular structure of corannulene. The differences between the experimental C–C bond lengths correlate to the double-bond character predicted using Kekule’s resonance structures. In the case of C20H10–H20, the water molecule is found to reside inside the bowl-like structure of corannulene. Our experimental and theoretical results indicate that the water molecule rotates freely around its C2 axis and that dispersion interactions are the dominant contribution to the binding.
“Prediction of the rotational spectra of microsolvated complexes with low cost DFT methods”
P. Pinacho, J.C. López, S. Blanco*
J. Mol. Spectrosc., 337, (2017), 145-152. DOI: 10.1016/j.jms.2017.02.018
Some of the most used DFT methods together with MP2 have been tested using 6-311++G(d,p) and TZVP basis sets to probe their usefulness in prediction of the rotational spectra of several microsolvated complexes of formamide, t-N-methylformamide, glycine and b-propiolactone. Results obtained for the rotational parameters and the prediction of the spectra have been compared to experimental data previously measured by Fourier Transform Microwave Spectroscopy. Analysis of the standard deviation of the predicted rotational spectra for all the levels tested indicates that the methods which better approach to the MP2 results are mPW91LYP and B3LYP-D3.
“The equivalence of the methyl groups in puckered 3,3-dimethyloxetane “
A. Macario, S. Blanco, J.C. López*
J. Mol. Spectrosc., 337, (2017), 72-79. DOI: 10.1016/j.jms.2017.04.004
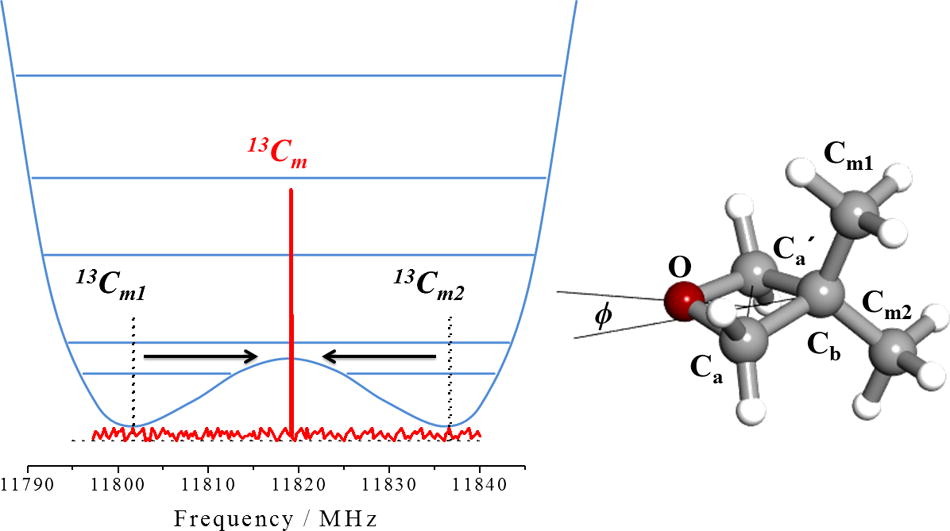
3,3-dimethyloxetane (DMO) has a nonplanar ring equilibrium configuration (f= 17.5º) and a double minimum potential function, with a barrier of 47 cm_1, for ring-puckering vibration. In a previous work, the observation of endocyclic 13C and 18O monosubstituted isotopologues allowed to conclude that the ring is puckered, consistent with the observed ring-puckering potential function. However, an interesting feature was observed for the 13C substitutions at the methyl carbon atoms. While two different, axial and equatorial, 13C-methyl groups spectra are predicted from a rigid nonplanar ring DMO model, only one spectrum was found. This behavior evidences that the two methyl groups of DMO are equivalent as could be expected for a planar ring. In this work we show how consideration of the potential function and the dynamical path for ring-puckering motion allows to reproduce the experimental results.
“Butterfly motion of formic acid and cyclobutanone in the 1:1 hydrogen bonded molecular cluster”
L. Evangelisti, L. Spada, W. Li, S. Blanco, J.C. López, A. Lesarri*, J.-U. Gravow, W. Caminati*
Phys. Chem. Chem. Phys., 19, (2017), 204-209. DOI: 10.1039/c6cp06941j
Upon supersonic expansion, formic acid and cyclobutanone (CBU) form a molecular cluster in which the two constituent molecules, linked by OH···O and CH···O hydrogen bonds, undergo a rapid interconversion between two equivalent forms. The tunneling motion takes place through the rupture and reformation of the C–H···O hydrogen bond between the carbonyl oxygen of HCOOH and one of the two hydrogen atoms of the methylenic group adjacent to the cyclobutanone keto group. From the microwave spectra, tunneling energy splittings (ΔE01) have been determined for the parent (1122.756(3) MHz), DCOOH···CBU (1084.538(1) MHz) and HCOOD···CBU (1180.282(4) MHz) isotopic species. From these splittings, the potential barrier to interconversion has been calculated to be B2 = 39.7(5) cm_1. The tunneling pathway is an asymmetric butterfly-like motion between the two moieties of the adduct, with a barrier at a configuration in which the ring plane of cyclobutanone is coplanar with formic acid.
2016
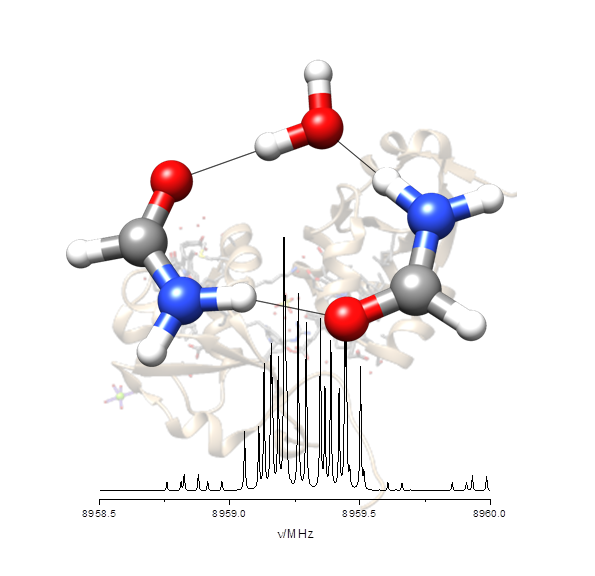
“Hydrogen-Bond Cooperativity in Formamide2-water: A Model for Water-Mediated Interactions”
S. Blanco, P. Pinacho, J.C. López*
Angew. Chem. Int. Ed., 55, (2016), 9331-9335. DOI: 10.1002/anie.201603319
Isolated formamide2–water can be taken as a simple model for water-mediated interactions and amide self-association. The structure and the electric field gradient determined at the nitrogen nuclei measured from the rotational spectrum reveal the subtle inductive effects that polarize the amide group and alter the C=O and N−C distances of formamide.
“Water-Induced Structural Changes in Crown Ethers from Broadband Rotational Spectroscopy”
C. Pérez, J.C. López*, S. Blanco, M. Schnell*
J. Phys. Chem. Letts., 7, (2016), 4053-4058. DOI: 10.1021/acs.jpclett.6b01939
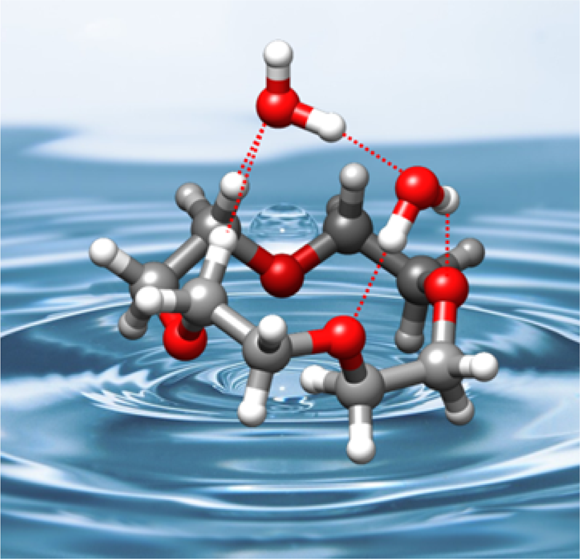
The complexes of 12-crown-4 ether (12C4) with water, generated in a supersonic jet, have been studied using broadband Fourier transform microwave spectroscopy. Three 1:1 and one 1:2 clusters have been observed and their structures unambiguously identified through the observation of isotopologue spectra. The structures of the clusters are based on networks of O−H···O and C−H···O hydrogen bonds. The most abundant 1:1 cluster is formed from the most stable S4 symmetry conformer of 12C4, even though it is not the energetically favored water complex. Interestingly, the structures of the most stable water cluster and the other remaining observed 1:1 and 1:2 complexes are formed from the second or the fifth most abundant conformers of 12C4. This shows the existence of a mechanism that changes the conformation of 12C4 so that the host−guest interactions can be maximized, even for a “soft” ligand like water.
“Wetting Camphor: Multi-Isotopic Substitution Identifies the Complementary Roles of Hydrogen Bonding and Dispersive Forces”
C. Pérez, A. Krin, A.L. Steber, J.C. López, Z. Kisiel, M. Schnell*
J. Phys. Chem. Letts., 7, (2016), 154-160. DOI: 10.1021/acs.jpclett.5b02541
Using broadband rotational spectroscopy, we report here on the delicate interplay between hydrogen bonds and dispersive forces when an unprecedentedly large organic molecule (camphor, C10H16O) is microsolvated with up to three molecules of water. Unambiguous assignment was achieved by performing multi H218O isotopic substitution of clustered water molecules. The observation of all possible mono- and multi-H218O insertions in the cluster structure yielded accurate structural information that is not otherwise achievable with single-substitution experiments. The observed clusters exhibit water chains starting with a strong hydrogen bond to the C=O group and terminated by a mainly van der Waals (dispersive) contact to one of the available sites at the monomer moiety. The effect of hydrogen bond cooperativity is noticeable, and the O···O distances between the clustered water subunits decrease with the number of attached water molecules. The results reported here will further contribute to reveal the hydrophobic and hydrophilic interactions in systems of increasing size.